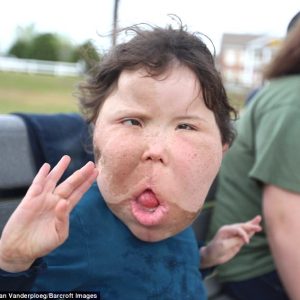
Apert syndrome, named after the French physician Eugene Apert who first described it in 1906, is a rare congenital disorder characterized by distinctive craniofacial and limb abnormalities. This condition arises from mutations in the fibroblast growth factor receptor 2 (FGFR2) gene, which plays a crucial role in the development of bones and tissues during fetal development.
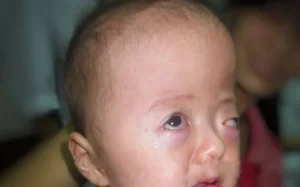
Individuals with Apert syndrome typically exhibit craniosynostosis, a condition in which the skull bones fuse prematurely, leading to an abnormal head shape. This often results in a prominent forehead, wide-set eyes (hypertelorism), a flat midface, and a beaked nose. Additionally, individuals may have syndactyly, where certain fingers or toes are fused together, particularly the second, third, and fourth digits. Other features of Apert syndrome may include dental abnormalities, hearing loss, and cognitive impairment, although the severity of these manifestations can vary widely among affected individuals.
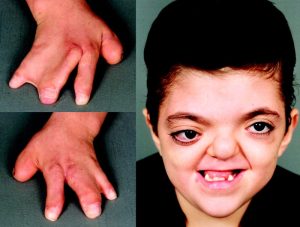
The diagnosis of Apert syndrome is usually based on clinical features observed at birth or during infancy. Imaging studies such as X-rays and CT scans may be performed to evaluate the extent of craniosynostosis and limb abnormalities. Genetic testing can confirm the presence of mutations in the FGFR2 gene, although this may not be necessary for diagnosis in all cases.
Management of Apert syndrome typically involves a multidisciplinary approach coordinated by a team of healthcare professionals, including craniofacial surgeons, orthopedic surgeons, pediatricians, and other specialists. Early intervention is essential to address craniosynostosis and prevent complications such as increased intracranial pressure and developmental delays. Surgical procedures to release fused skull bones (cranial vault reconstruction) and separate fused fingers or toes (syndactyly release) may be performed to improve function and appearance.

In addition to surgical interventions, individuals with Apert syndrome may require ongoing medical care to address associated complications such as hearing loss, dental problems, and vision impairment. Physical and occupational therapy may also be beneficial in promoting optimal development and maximizing functional abilities.
While there is currently no cure for Apert syndrome, advances in medical care and surgical techniques have greatly improved outcomes for affected individuals. With early diagnosis, comprehensive treatment, and ongoing support, many individuals with Apert syndrome can lead fulfilling lives and achieve their full potential.

However, living with Apert syndrome can present unique challenges, both for individuals affected by the condition and their families. Support groups and resources are available to provide information, guidance, and emotional support to those impacted by Apert syndrome.
In conclusion, Apert syndrome is a rare genetic disorder characterized by craniofacial and limb abnormalities resulting from mutations in the FGFR2 gene. While the condition presents significant challenges, early intervention and comprehensive care can greatly improve outcomes and enhance the quality of life for affected individuals. Continued research into the underlying mechanisms of Apert syndrome and advancements in treatment modalities offer hope for the future management of this complex condition.